
우선 정식 튜토리얼은 다음과 같습니다.
- http://www.incodom.kr/Seurat#h_8bb7bcd86febfdc9813f69cc4a6d4c78
- https://scrnaseq-course.cog.sanger.ac.uk/website/seurat-chapter.html
library(dplyr)
install.packages("Seurat")
library(Seurat)
### import input_data(cellranger count output과 동일) ###
pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
### setup the seurat object ###
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
### Preprocessing(QC) ###
### Low quality cell, mitochondria genome percent check
pbmc[["percent.mt"]] <- PercentageFeatureSet(object = pbmc, pattern = "^MT-")
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 15) # quality control
### normalizing the data ###
pbmc <- NormalizeData(object = pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
### identification of highly variable features ###
pbmc <- FindVariableFeatures(object = pbmc, selection.method = "vst", nfeatures = 2000)
### scaling data ###
all.genes <- rownames(x = pbmc)
pbmc <- ScaleData(object = pbmc, features = all.genes)
### perform linear dimensional reduction ###
pbmc <- RunPCA(object = pbmc, features = VariableFeatures(object = pbmc))
### determine the "dimensionality" of the dataset
pbmc <- JackStraw(object = pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(object = pbmc, dims = 1:20)
### cluster the cells ###
pbmc <- FindNeighbors(object = pbmc, dims = 1:10)
pbmc <- FindClusters(object = pbmc, resolution = 0.5)
### Run non-linear dimensional reduction(tSNE) ###
pbmc_umap <- RunUMAP(object = pbmc, dims = 1:10)
DimPlot(object = pbmc_umap, reduction = "umap")
pbmc_tsne <- RunTSNE(object = pbmc, dims = 1:10)
DimPlot(object = pbmc_tsne, reduction = "tsne")
### Finding differentially expressed features(biomarkers) ###
pbmc.markers_umap <- FindAllMarkers(object = pbmc_umap, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
pbmc.markers_tsne <- FindAllMarkers(object = pbmc_tsne, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
### Maybe you need this kind of data ###
umap <- DimPlot(object = pbmc_umap, reduction = "umap")
names(umap)
# [1] "data" "layers" "scales" "mapping" "theme"
# [6] "coordinates" "facet" "plot_env" "labels" "guides"
tsne <- DimPlot(object = pbmc_tsne, reduction = "tsne")
write.csv(umap$data, "C:/Users/sun/Desktop/RESULT.csv")
write.csv(tsne$data, "C:/Users/sun/Desktop/result.csv")
write.csv(pbmc.markers_umap, "C:/Users/sun/Desktop/markers_umap.csv")
write.csv(pbmc.markers_tsne, "C:/Users/sun/Desktop/markers_tsne.csv")다음은 위 코드에 대한 해석입니다.
- scRNA-seq data는 read count를 총 transcript로 나눈 뒤 10,000을 곱한 것을 나타냅니다.
- CreateSeuratObject에서 min.features 값이 작아야 누락되는 데이터가 작아집니다.
- FindVariableFeatures 함수에서 nfeatures 파라미터를 통해 가장 발현률이 높은 2,000개의 유전자들이 선택됩니다. nfeatures 값이 커야 클러스터링이 잘 되며 일반적으로 2,000 이상이면 클러스터링에 큰 영향을 주지 않습니다.
- NormalizeData를 통해 scRNA-seq data는 log-normalization 됩니다. 그 뒤 데이터는 total cellular read count와 mitochondrial read count의 회귀를 통해 z-score로 스케일링 됩니다.
- RunPCA를 통해 가장 유명한 차원 축소 알고리즘인 PCA(principal component analysis) 알고리즘이 실행됩니다. 이는 이후의 과정에서 클러스터링의 시간을 단축하고 computing burden을 줄이기 위함입니다. 제가 실행했을 때는 유전자 개수 만큼의 주성분 중에서 50개의 주성분이 클러스터링에 활용됐습니다.
- JackStraw와 ScoreJackStraw는 dimensionality를 결정하기 위함으로 이미 레퍼런스 상에 어떤 파라미터를 쓰라고 정해줬으면 그 부분을 생략할 수 있습니다. 따라서 이후의 파라미터가 동일하다면 num.replicate가 1이든 100이든 추후 진행에 차이가 없습니다.
- FindClusters를 통해 클러스터링을 진행하는 데 일반적으로 0.5의 resolution이 사용됩니다. resolution 값이 커야 cell type을 더 다양하게 분류해 줍니다. FindClusters는 graph-based approach를 사용합니다.
- RunUMAP에서 dims를 통해 다소 개형을 바꿀 수 있습니다.
- FindAllMarkers에서는 기본적으로 Wilcoxon rank sum test를 사용하는 데 이 밖에도 다른 통계기법을 사용할 수 있습니다. 주어진 파라미터 셋팅에서는 각 클러스터 내 25% 이상의 cell에서 발현하고 있고 fold change가 0.25 (log scale)보다 커야 marker gene으로 취급합니다. 단, only.pos가 TRUE이기 때문에 fold change가 양수인 유전자만 도출됩니다.
- 똑같은 코드를 쓰면 똑같은 결과가 나오지만 컴퓨터 종류에 따라 결과가 다르게 나올 때도 있고 같게 나올 때도 있습니다. 초기 조건이 약간 차이가 나는 듯 합니다.
- Dot Plot은 다음을 참고합니다: https://github.com/satijalab/seurat/issues/2491
- Feature Plot은 다음을 참고합니다: https://github.com/satijalab/seurat/issues/2400
이제 주어진 데이터를 이해할 필요가 있는데요. 주어진 데이터는 filtered_gene_bc_matrices/hg19/ 디렉토리 안에 총 세 개의 파일로 저장돼 있습니다.
- barcodes.tsv : 각 single cell들의 바코드 네임
- genes.tsv : 유전자의 정식 명칭과 별명
- matrix.mtx : 각 바코드와 각 유전자, 각 유전자의 발현량을 sparse matrix로 나타낸 것
참고로 matrix.mtx는 다음과 같은 sparse matrix입니다.
- 0이 아닌 각 데이터가 나타내는 유전자가 genes.tsv에서 몇 번째인지를 첫 번째 값이라고 합시다.
- 0이 아닌 각 데이터가 나타내는 single cell의 바코드가 무엇인지를 두 번째 값이라고 합시다.
- 0이 아닌 각 데이터에 표시된 발현량을 세 번째 값이라고 합시다.
- 첫 번째, 두 번째, 세 번째 값을 한 행으로 하여 행들을 쌓아서 sparse matrix를 구성합니다.
- 전체 행렬을 텍스트 파일로 저장하고 .mtx 파일로 변환합니다.
- 처음에 다음과 같이 세 줄을 추가합니다. 참고로 32,738은 유전자의 개수, 1,047은 바코드의 개수, 2,572,919는 0이 아닌 데이터의 개수입니다.
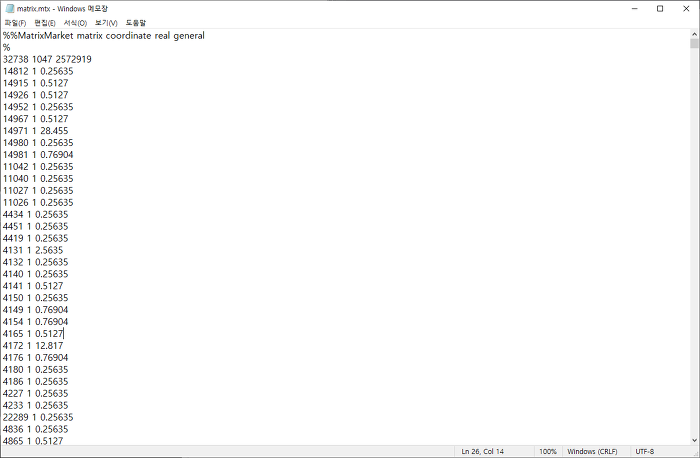
자신의 데이터로 분석을 하고 싶은 경우 Read10X로 읽어들인 pbmc.data에서 행렬 다루듯이 수정합니다.
다음은 주요 트러블슈팅입니다.
문제점 1. "파일이 너무 커서 메모장에서 열 수 없습니다."
텍스트 파일이 1 GB 근처를 넘기 시작하면 메모장을 통해 위와 같은 내용을 읽어보기가 거의 불가능합니다. 워드패드는 너무 오래 걸리는 데다 제대로 원하는 기능을 수행하지 못했습니다. gVim 8.0 등과 같은 프로그램을 통해 용량이 상당히 큰 텍스트 파일을 편집할 수 있습니다.
문제점 2. "readMM(): column values 'j' are not in 1:nc"
matrix.mtx에 표기된 바코드 순서가 barcodes.tsv의 바코드 개수의 범위를 넘어가면 위와 같은 오류 메시지를 출력합니다.
문제점 3. "Directory provided does not exist"
pbmc.data <- Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/")
# Error in Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/") :
# Directory provided does not exist파일을 직접 다운받아서 압축을 푼 뒤 생성된 문서의 디렉토리 주소를 바꿔주어야 합니다. 파일은 여러 사이트에 공개돼 있습니다. 그러고선 다음과 같이 수정해 주면 됩니다.
pbmc.data <- Read10X(data.dir = "C:/Users/sun/Desktop/filtered_gene_bc_matrices/hg19/")문제점 4. "에러: package or namespace load failed for ‘Seurat’ in loadNamespace(j <- i[[1L]], c(lib.loc, .libPaths()) versionCheck = vI[[j]]): ‘multtest’이라고 불리는 패키지가 없습니다"
install.packages("BiocManager")
BiocManager::install('multtest')
install.packages("Seurat")
library(Seurat)문제점 5. "에러: 크기가 13.3 GB인 벡터를 할당할 수 없습니다"
memory.size(max = TRUE) # OS로부터 R이 사용 가능한 메모리
memory.size(max = FALSE) # 현재 사용중인 메모리 크기
memory.limit(size = NA) # 컴퓨터의 최대 메모리 한계치
memory.limit(size = 50000) # 컴퓨터의 최대 메모리 한계치 약 49GB로 높이기작업관리자 → 성능 → 메모리를 들어가시면 총 메모리의 용량이 얼마인지 알 수 있습니다. 저의 경우 기본적으로 Seurat를 돌리는 데 7 GB를 잡아먹고 데이터 크기에 따라 추가적인 메모리를 요구합니다. (예 : 13.3 GB) 사용된 슬롯을 사용하고 추가로 램을 구입하는 게 하나의 방법이 될 수 있습니다. 저는 64 GB 램을 쓰고 있고 데이터 분석을 한다고 한다면 32 GB 이상은 돼야 한다고 생각합니다.
일반적으로 pbmc ← ScaleData(object = pbmc, features = all.genes) 부분에서 메모리 수요가 큽니다. pbmc ← JackStraw(object = pbmc, num.replicate = 100) 부분에서도 수요가 큰데 num.replicate = 1로 바꾸면 됩니다. 한편, memory.limit으로 늘릴 수 있는 메모리는 PC의 램의 메모리를 초과할 수 없다는 점은 중요합니다.
문제점 6. 입력 파일이 .mtx 파일이 아니라 .rds 파일인 경우
library(dplyr)
library(Seurat)
### read & write ###
data <- readRDS("C:/Users/sun/Desktop/GSM4557327_555_1_cell.counts.matrices.rds", refhook = NULL)
names(data)
# [1] "exon" "intron" "spanning"
saveRDS(data, "C:/Users/sun/Desktop/Copy.rds", ascii = FALSE, version = NULL, compress = TRUE, refhook = NULL)
### barcodes ###
head(colnames(data$exon))
# [1] "TTGCTAAGCAGT" "AACGACGGGTCT" "AACGGGGGCGAG" "CAGCAGAGGGTC"
# [5] "CAAGAACACTAT" "CCCCTTATGTCG"
### genes ###
head(rownames(data$exon))
# [1] "KLK11" "CCDC159" "C7orf50" "RP11-1437A8.4"
# [5] "EMP2" "IFI27L2"
pbmc.data <- data$exon
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc[["percent.mt"]] <- PercentageFeatureSet(object = pbmc, pattern = "^MT-")
pbmc <- NormalizeData(object = pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- FindVariableFeatures(object = pbmc, selection.method = "vst", nfeatures = 2000)
all.genes <- rownames(x = pbmc)
pbmc <- ScaleData(object = pbmc, features = all.genes)
pbmc <- RunPCA(object = pbmc, features = VariableFeatures(object = pbmc))
pbmc <- JackStraw(object = pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(object = pbmc, dims = 1:20)
pbmc <- FindNeighbors(object = pbmc, dims = 1:10)
pbmc <- FindClusters(object = pbmc, resolution = 0.5)
pbmc <- RunUMAP(object = pbmc, dims = 1:10)
DimPlot(object = pbmc, reduction = "umap")문제점 7. "dimnamesGets(x, value)에서 다음과 같은 에러가 발생했습니다: invalid dimnames given for “dgTMatrix” object"
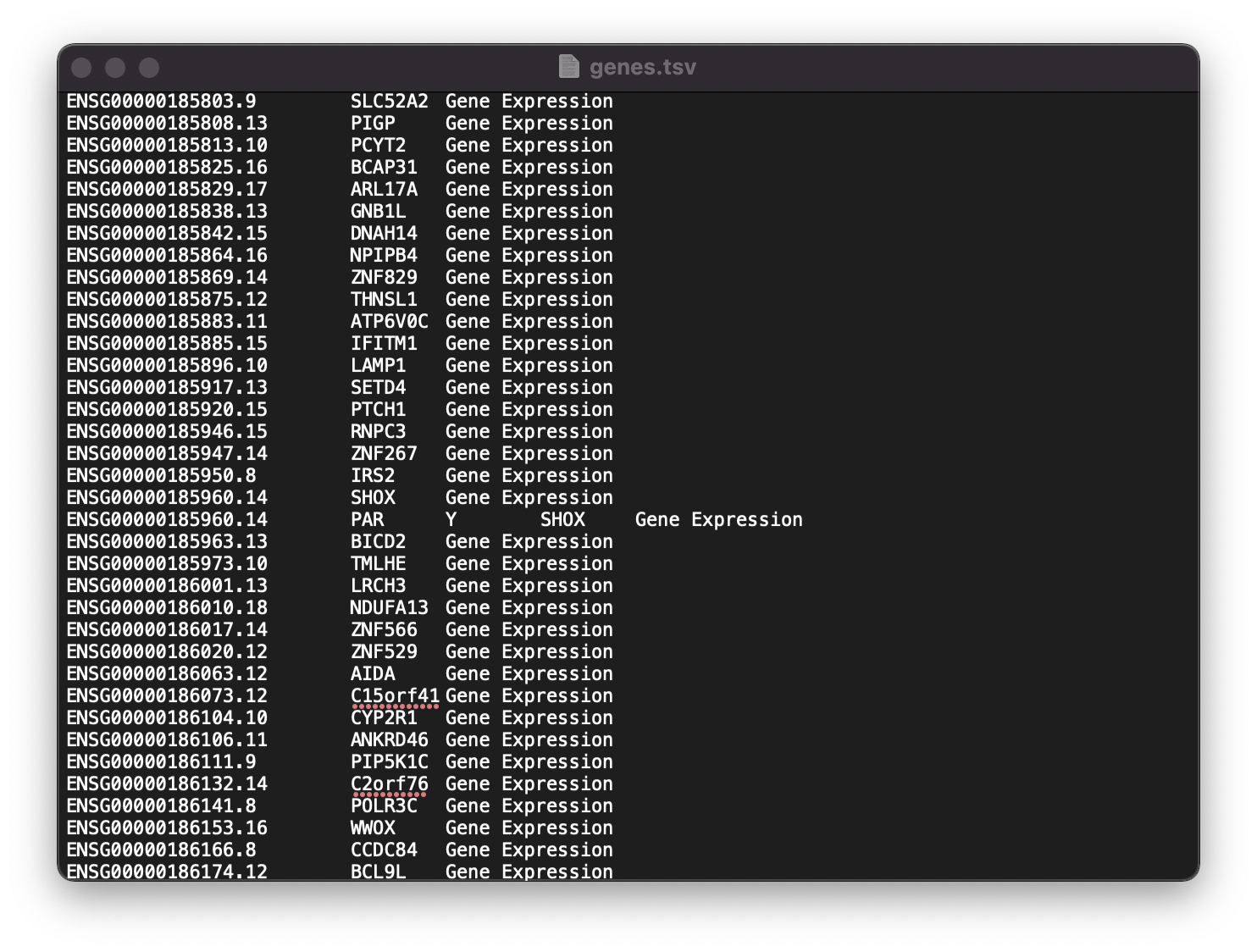
이 문제는 위와 같이 genes.tsv에서 값들이 잘 정렬되지 않아 Read10X 부분에서 문제를 일으키는 것입니다. 모든 tuple들이 3열이 되도록 잘 정리한다면 다음 단계가 smooth하게 진행될 것입니다.
이상 포트래이 tech 블로그였습니다.




